Dataset for "Optimising Drug Activity Using Docking-Informed Machine Learning"
This data set contains the structures and numerical features of molecules known to be active or inactive against a range of drug target proteins. They also include measurements of their true activity values from existing databases, as well as estimated activity values from simple computer-based docking simulations. Code used to obtain and process this data is provided, as well as scripts used to perform Bayesian optimisation (BO) experiments on the completed data sets.
This data archive includes files for the journal article titled "Optimising Drug Activity Using Docking-Informed Machine Learning". The archive contains data sets of molecular structures (represented as SMILES strings), numerical descriptors, pre-processed activity values and molecular docking scores for known actives and inactives against a number of different protein targets. This includes data for 14 targets derived from the ChEMBL database (EGFR, JAK2, LCK, MAOB, NOS1, PARP1, ACHE, PDE5A, PTGS2, ESR1, NR3C1, AR, F10, ADRB2), 4 targets from LIT-PCBA datasets (ESR1ago, ESR1ant, PAPRG, TP53), and 4 targets (DRD2, USP7, Mpro, TYK2) derived from a dataset used in a different research article for benchmarking active learning (BAL). Additionally, the docked poses of compounds and the corresponding pre-processed protein structures used for docking are available in this archive. Python scripts that were used to obtain and process this data, and to perform molecular docking, are also provided. The Python scripts used to perform and analyse the Bayesian optimisation (BO) machine learning experiments are also available in this archive, in addition to the bash command line scripts used to submit these experiments. Some of these scripts are specific to a given source (for the ChEMBL, LIT-PCBA and BAL data sets), while some are generic (for example, scripts used to analyse or plot results).
Cite this dataset as:
Proudfoot, J.,
Lewis-Atwell, T.,
Grayson, M.,
2026.
Dataset for "Optimising Drug Activity Using Docking-Informed Machine Learning".
Bath: University of Bath Research Data Archive.
Available from: https://doi.org/10.15125/BATH-01496.
Export
Data
EGFR.tar.gz
application/gzip (18MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target EGFR (epidermal growth factor receptor).
JAK2.tar.gz
application/gzip (21MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target JAK2 (janus kinase 2).
LCK.tar.gz
application/gzip (13MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target LCK (tyrosin-protein kinase Lck).
MAOB.tar.gz
application/gzip (5MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target MAOB (monoamine oxidase B).
NOS1.tar.gz
application/gzip (3MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target NOS1 (nitric oxide synthase 1).
PARP1.tar.gz
application/gzip (12MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target PARP1 (poly [ADP-ribose] polymerase 1).
ACHE.tar.gz
application/gzip (15MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target ACHE (acetylcholineesterase).
PDE5A.tar.gz
application/gzip (10MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target PDE5A (phosphodiesterase 5A).
PTGS2.tar.gz
application/gzip (8MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target PTGS2 (prostaglandin-endoperoxide synthase 2).
ESR1.tar.gz
application/gzip (15MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target ESR1 (estrogen receptor alpha).
NR3C1.tar.gz
application/gzip (15MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target NR3C1 (glucocorticoid receptor).
AR.tar.gz
application/gzip (9MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target AR (androgen receptor).
F10.tar.gz
application/gzip (51MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target F10 (coagulation factor X).
ADRB2.tar.gz
application/gzip (14MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the ChEMBL target ADRB2 (beta-2 adrenergic receptor).
ESR1ago.tar.gz
application/gzip (12MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the LIT-PCBA target ESR1ago (agonists of ESR1; estrogen receptor alpha).
ESR1ant.tar.gz
application/gzip (12MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the LIT-PCBA target ESR1ant (antagonists of ESR1; estrogen receptor alpha).
PPARG.tar.gz
application/gzip (12MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the LIT-PCBA target PPARG (peroxisome proliferator-activated receptor gamma).
TP53.tar.gz
application/gzip (10MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the LIT-PCBA target TP53 (tumor protein P53).
DRD2.tar.gz
application/gzip (28MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the BAL target DRD2 (dopamine D2 receptor).
USP7.tar.gz
application/gzip (56MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the BAL target USP7 (Ubiquitin-specific-processing protease 7).
Mpro.tar.gz
application/gzip (6MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the BAL target Mpro (SARS-CoV-2 main protease).
TYK2.tar.gz
application/gzip (8MB)
Creative Commons: Attribution-Share Alike 4.0
Data for the BAL target TYK2 (tyrosine kinase 2)
Code
ChEMBL_scripts.tar.gz
application/gzip (14kB)
Software: GNU GPL 3.0+
Python scripts for creating the ChEMBL data sets and performing Bayesian optimisation.
LIT-PCBA_scripts.tar.gz
application/gzip (5kB)
Software: GNU GPL 3.0+
Python scripts for creating the LIT-PCBA data sets and performing Bayesian optimisation.
BAL_scripts.tar.gz
application/gzip (12kB)
Software: GNU GPL 3.0+
Python scripts for creating the BAL data sets and performing Bayesian optimisation.
Analysis_scripts.tar.gz
application/gzip (12kB)
Software: GNU GPL 3.0+
Python scripts for analysing the data sets and Bayesian optimisation results.
Github repository for the code used in the experiments described in the journal article.
Creators
James Proudfoot
University of Bath
Toby Lewis-Atwell
University of Bath
Matt Grayson
University of Bath
Contributors
University of Bath
Rights Holder
Coverage
Collection date(s):
From March 2024 to October 2025
Documentation
Data collection method:
For ChEMBL targets, 14 representative targets were selected from DOCKSTRING: 3 kinases (EGFR, JAK2, LCK), 6 other enzymes (MAOB, NOS1, ACHE, PARP1, PTGS2, PDE5A), 3 nuclear receptors (ESR1, NR3C1, AR), and 2 other proteins (ADRB2, F10). Ligand SMILES and activity data (Ki: nanomolar equilibrium constant, nM) were obtained programmatically from ChEMBL (https://www.ebi.ac.uk/chembl/) via the ChEMBL webresource Python API. Ki values were log-scaled to pKi values (log-equilibrium constant, log-M) and duplicate entries (entries with the same canonical SMILES) were merged by taking the entry with the highest (max) pKi value. RDKit was used to generate machine learning features from ligand SMILES strings, including all available 2D RDKit features, MACCS (molecular access tokens), and 2048-bit radius-3 Morgan molecular fingerprints. This data was collected in the "{target}_data_pKi.csv" files. The DOCKSTRING Python package was used to dock all ligands to their respective target using AutoDock Vina (v1.2.6). In this package, ligand SMILES are protonated at pH 7.4 using OpenBabel, 3D geometries generated using RDKit ETKG atom-embedding and optimized with the MMFF94 force field, before being docked into the {target}.pdb file geometry with the docking cuboid (cartesian position, dimensions in Angstrom) specified by the {target}_conf.txt file. This produces an SDF file for each ligand in the {target}/conformers directory, labelled by its compound ChEMBL ID, containing 8 docked poses sorted by decreasing docking score magnitude. For the pose with the highest AutoDock Vina docking score, 3D RDKit descriptors were generated and stored. Docking scores and 3D descriptors were appended to the "{target}_data_pKi.csv" file to create the intermediate "{target}_data_3d_pKi.csv" (not contained in this data archive). The top-ranked docked poses were then re-scored with the ML-based Python program delta_LinF9_XGB, which was trained on large datasets of protein-ligand geometries and protein-ligand affinities. Intermediate vina features, ligand features, LinF9 docking score, and delta_LinF9_XGB docking scores were then appended to the "{target}_data_3d_pKi.csv" file. For each target, all ligands were clustered using the first 2 PCA or first 2 t-SNE components of the Morgan fingerprints, with k-means clustering from the SciKit-Learn Python package (k={10,20,50,100}), to create the final "{target}-2048_data_3d_pKi.csv dataset (labelled "2048" due to the length of the Morgan fingerprints). LIT-PCBA is an external benchmark dataset for testing virtual screening methods. For LIT-PCBA, the 4 smallest datasets (ESR1ago, ESR1ant, PPARG, TP53) were collected from the DOCKM8 Zenodo archive (lit-pcba.zip). DOCKM8 is an all-in-one Structure-Based Virtual Screening workflow for consensus docking (a method in molecular docking where multiple docking scores from different progams are ensembled by consensus to give a more accurate final score). Instead of performing docking ourselves, we used the docking scores obtained in the DOCKM8 research for these LIT-PCBA targets. We used the DOCKM8 GNINA docked poses ("{target}_poses.sdf") to generate RDKit 2D and 3D descriptors, and DOCKM8 GNINA docking scores ("{target}_scores.csv") as our docking scores for docking-informed machine learning (Bayesian) optimization. Ligand activities were obtained from PubChem Bioassays by manually downloading the activity datasets associated with the following assay IDs (ESR1ago: 743075, ESR1ant: 743080, PPARG: 743094, TP53: 651631), collected in the "{target}_datatable.csv" files. EC50 (or equivalent, e.g. AC50, IC50) values were log-scaled to pEC50 values (log-half-maximal effective concentration, log-M) and duplicate entries (entries with the same canonical SMILES) were merged by taking the mean of activity. Ligand SMILES, ligand 2D/3D RDKit descriptors, activity values and docking scores were collected in a final CSV file "{target}_data_full.csv". All ligands were again clustered using the same methodology as for ChEMBL datasets, updating the final CSV file with columns identifying the cluster each ligand is associated with. For the BAL data sets, data sets containing molecular structures (SMILES) and activity endpoints (pIC50 or pKi values) were collected from the GitHub repository associated with the paper "Benchmarking active learning protocols for ligand-binding affinity prediction" by Gorantla et al. All compounds were featurised in the same way as for the ChEMBL datasets (creating Morgan fingerprints and 2D descriptors using RDKit), followed by clustering on fingerprints. For the DRD2, USP7 and Mpro data sets, compounds were docked against representative PDB structures of the protein targets from the protein data bank (IDs: 9BS9, 5N9T, 9ZNL, respectively), which were converted to PDBQT files by protonation with PropKa via PDB2PQR, followed by protein preparation with prepare_receptor4.py from MGLTools. For the TYK2 data set, docked poses and docking scores from Glide were obtained from the Zenodo archive associated with the original data source (10.5281/zenodo.13759489), and re-scoring was performed against a processed structure of the TYK2 receptor derived from the 4GIH PDB entry.
Data processing and preparation activities:
Local changes to the DOCKSTRING, delta_LinF9_XGB and MGLTools software packages were made. These local versions can be found in the GitHub repository associated with this dataset (https://github.com/the-grayson-group/finding_ligands) and the changes are outlined in the README.md file. DOCKM8 docking scores were collected and merged with ligand SMILES, ligand 2D/3D RDKit descriptors, and PubChem pEC50 activity values, and duplicate ligand entries were merged as outlined above.
Technical details and requirements:
Schrödinger Maestro viewer (free academic license - project not associated with commercial partner) was used to view protein and ligand 3D geometries from PDB/SDF files. OpenBabel (v3.1.1) must be downloaded separately (http://openbabel.org/docs/Installation/install.html) before creating Python environments. Python3 and Anaconda v24.9.2 were used to create the Python environments for the majority of the data collection used in this project. The environment YAML files can be found in the associated GitHub repository, and the conda environments can be re-created using the steps outlined in the README.md file. Local versions of the DOCKSTRING, delta_LinF9_XGB and MGLTools packages can be found in the associated GitHub repository. The version of the GitHub repository associated with this data archive is Release v1.0.0 (https://github.com/the-grayson-group/finding_ligands/releases/tag/v1.0.0).
Additional information:
"{target}" refers to the gene code for the various protein targets investigated in this project; 14 ChEMBL targets (EGFR, LCK, JAK2, MAOB, NOS1, PARP1, ACHE, PDE5A, PTGS2, ESR1, NR3C1, AR, F10, ADRB2), 4 LIT-PCBA targets (ESR1ago, ESR1ant, PPARG, TP53) and 4 BAL targets (DRD2, USP7, Mpro, TYK2). Each data set is of the form {target}.tar.gz, and contains folders, which themselves contain data files. For ChEMBL targets, the main data files are CSV files marked as "{target}_data_pKi.csv.", and "{target}-2048_data_3d_delta_pKi.csv.". They contain ligand SMILES, activities, activity values (processed pKi values derived from ChEMBL v34 records), 2D RDKit features (including 2048-bit radius-3 Morgan fingerprints), and the latter additionally contains 3D RDKit features, AutoDock Vina features and docking scores (including AutoDock Vina, LinF9, delta_LinF9_XGB docking scores). The “conformers” folders contain SDF and PDB files of 3D ligand poses docked to the respective "{target}_target.pdbqt" PDBQT file using AutoDock Vina (v1.2.6) (with the AutoDock Vina docking cell defined by the "{target}_conf.txt" file). Files labelled "{target}.pdb" are PDB files used for docking re-scoring. For LIT-PCBA targets, CSV files marked "{target}_data_full.csv" contain ligand SMILES, activity values (processed pEC50 values derived from PubChem BioAssays records), all GNINA CNN-Affinity docking scores, 2D RDKit features, and 3D RDKit features of docked poses. For BAL targets, CSV files marked "{target}_data_3d_delta_{activity}.csv" contain ligand SMILES, activity values (processed activity values, either pIC50, or pKi values derived from the BAL data set GitHub repository), all docking scores, 2D RDKit features, and 3D RDKit features of docked poses. The “conformers” folders contain SDF and PDB files of 3D ligand poses docked to the respective "{target}_target.pdbqt" PDBQT file. Files labelled "{target}.pdb" are PDB files used for docking re-scoring. The compressed files for each dataset "{dataset}_scripts.tar.gz" contain the key python files used to either obtain or process data from external sources, to featurise molecules by creating numerical descriptors, scripts used to perform molecular docking and re-scoring, and code used to run Bayesian optimisation (BO) experiments, including bash submit scripts. Note: the ChEMBL database is continuously updated as new data is collected from new data sources (e.g. published medicinal chemistry journal articles). Therefore, using the current script to collect ChEMBL ligands and activities from the Python ChEMBL webresource client API will not necessarily reproduce the "{target}_data_pKi.csv" files exactly. For the purposes of replicating the results of this project, start with the "{target}_data_pKi.csv" contained in this repository or the associated GitHub repository, instead of generating them directly using the Python scripts found in the GitHub repository. Alternatively, download a local version of ChEMBL v34 and generate the initial data sets of compounds and pKi values from the local version. The original data was collected from ChEMBL v34, March 2024.
Methodology link:
García-Ortegón, M., Simm, G. N. C., Tripp, A. J., Hernández-Lobato, J. M., Bender, A., and Bacallado, S., 2023. DOCKSTRING. GitHub. Available from: https://github.com/dockstring/dockstring.
Yang, C., 2022. Delta_LinF9_XGB. GitHub. Available from: https://github.com/dockstring/dockstring.
Lacour, A., Volkamer, A., Hirsch, A., and Ibrahim, H., 2024. DockM8_Benchmarking_results. Version v1.0.0. Zenodo. Available from: https://doi.org/10.5281/ZENODO.11191685.
Lacour, A., Volkamer, A., Hirsch, A., and Ibrahim, H., 2024. DockM8. GitHub. Available from: https://github.com/DrugBud-Suite/DockM8.
Gorantla, R., 2023. Benchmarking Active Learning Protocols for Ligand Binding Affinity Prediction. GitHub. Available from: https://github.com/meyresearch/ActiveLearning_BindingAffinity.
Google Research, 2022. Active Learning for Free Energy Perturbation. GitHub. Available from: https://github.com/google-research/google-research/tree/master/al_for_fep.
Documentation Files
README.md
text/plain (7kB)
Creative Commons: Attribution-Share Alike 4.0
ReadMe file from the associated GitHub repository. Can be viewed in Markdown format in the repository itself.
plot.png
image/png (2MB)
Creative Commons: Attribution-Share Alike 4.0
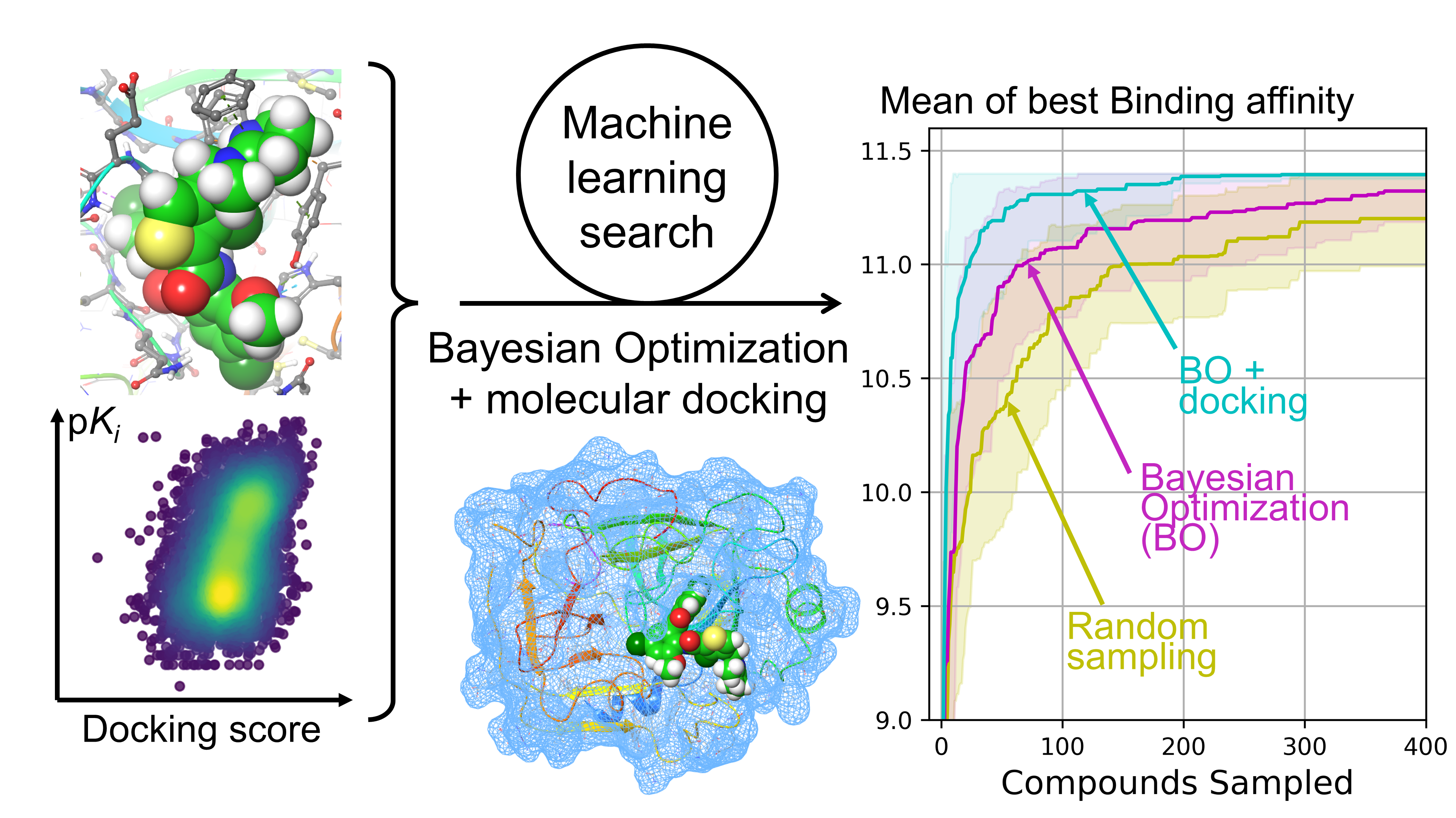
Graphical abstract from the associated GitHub repository.
Funders
UK Research and Innovation
https://doi.org/10.13039/100014013
UKRI Centre for Doctoral Training in Accountable, Responsible and Transparent AI
EP/S023437/1
Publication details
Publication date: 13 July 2026
by: University of Bath
Version: 1
DOI: https://doi.org/10.15125/BATH-01496
URL for this record: https://researchdata.bath.ac.uk/1496
Related datasets and code
Blackshaw, J., Adasme Mora, M. F., Arcila Toro, R., Bosc, N., Corbett, S., De Veij, M., Félix, E., Hunter, F., Ioannidis, H., Kizilören, T., Leach, A., Manners, E., Margariños, M. P., Mendez Lopez, D., Mosquera Morales, J. F., Smit, I., and Zdrazil, B., 2009. CHEMBL database release 34. In: ChEMBL Database. EMBL-EBI. Available from: https://doi.org/10.6019/chembl.database.34.
Contact information
Please contact the Research Data Service in the first instance for all matters concerning this item.
Contact person: Matt Grayson
Faculty of Science
Chemistry
Research Centres & Institutes
UKRI CDT in Accountable, Responsible and Transparent AI

{kind=link}