Dataset for "Comparing the Performances of Force Fields in Conformational Searching of Hydrogen Bond-Donating Catalysts"
This dataset contains all of the fully optimised structures (from force field conformational searches and DFT) used in the work titled "Comparing the Performances of Force Fields in Conformational Searching of Hydrogen Bond-Donating Catalysts". Twenty organic molecules were conformationally searched with eight force fields (OPLS3e, OPLS-2005, MMFF, MMFFs, AMBER*, OPLS, MM2* and MM3*) and all of the conformer structures were geometry optimised at the M06-2X/6-31G(d) level of theory and single-point energies of the resulting minima were calculated with the M06-2X/def2-TZVPP level of theory. This dataset contains the .mol2 files that correspond to the conformer structures found by the force fields, and the .out files for the DFT-optimised minima and the single-point energies. Also provided are .csv files containing all of the conformer energies according to the force fields and DFT.
Cite this dataset as:
Lewis-Atwell, T.,
Townsend, P.,
Grayson, M.,
2022.
Dataset for "Comparing the Performances of Force Fields in Conformational Searching of Hydrogen Bond-Donating Catalysts".
Bath: University of Bath Research Data Archive.
Available from: https://doi.org/10.15125/BATH-01065.
Export
Data
antilla.zip
application/zip (7MB)
Creative Commons: Attribution 4.0
bach.zip
application/zip (69MB)
Creative Commons: Attribution 4.0
berkessel.zip
application/zip (435MB)
Creative Commons: Attribution 4.0
boscalid.zip
application/zip (24MB)
Creative Commons: Attribution 4.0
cinchonidine.zip
application/zip (19MB)
Creative Commons: Attribution 4.0
gobel.zip
application/zip (74MB)
Creative Commons: Attribution 4.0
hexynoate.zip
application/zip (24MB)
Creative Commons: Attribution 4.0
inoue.zip
application/zip (117MB)
Creative Commons: Attribution 4.0
johnston.zip
application/zip (103MB)
Creative Commons: Attribution 4.0
mikami.zip
application/zip (128MB)
Creative Commons: Attribution 4.0
nakano.zip
application/zip (107MB)
Creative Commons: Attribution 4.0
ricci.zip
application/zip (56MB)
Creative Commons: Attribution 4.0
sasai.zip
application/zip (142MB)
Creative Commons: Attribution 4.0
segphos.zip
application/zip (34MB)
Creative Commons: Attribution 4.0
sulphon.zip
application/zip (83MB)
Creative Commons: Attribution 4.0
superquat.zip
application/zip (69MB)
Creative Commons: Attribution 4.0
takemoto.zip
application/zip (175MB)
Creative Commons: Attribution 4.0
terada.zip
application/zip (12MB)
Creative Commons: Attribution 4.0
urea_cinchona.zip
application/zip (280MB)
Creative Commons: Attribution 4.0
yamamoto.zip
application/zip (97MB)
Creative Commons: Attribution 4.0
csvs.zip
application/zip (181kB)
Creative Commons: Attribution 4.0
Creators
Toby Lewis-Atwell
University of Bath
Piers Townsend
University of Bath
Matthew Grayson
University of Bath
Contributors
University of Bath
Rights Holder
Documentation
Data collection method:
Conformational searches were performed with the force fields available in Schrödinger’s MacroModel v12.6 (OPLS3e, OPLS-2005, MMFF, MMFFs, AMBER*, OPLS, MM2* and MM3*) on each of 20 molecules. Aside from the force field potentials, the other settings used in the conformational searches were as follows: solvent was set to “None” (gas-phase); the energy minimization method was set to “Polak-Ribiere Conjugate Gradient” (PRCG), converging on the gradient with convergence threshold set to 0.001; the search method was set to “Mixed torsional/Low-mode sampling” with the energy window for saving structures set to 50 kJ mol-1 and the maximum atom deviation for structures to be considered different conformers was set to 0.25 Å. All DFT calculations were performed using the Gaussian 16, Revision A.03 software. Geometry optimizations were performed on all conformers of all molecules at the M06-2X/6-31G(d) level of theory in the IEFPCM(benzene) solvent model. Once all of the conformer structures had reached minima and were stationary points, single-point energies were calculated at the M06-2X/def2-TZVPP IEFPCM(benzene) level of theory. This level of theory was chosen since it has previously been used with success in previous reaction modelling studies.
Technical details and requirements:
Schrödinger MacroModel v12.6; Gaussian 16, Revision A.03
Additional information:
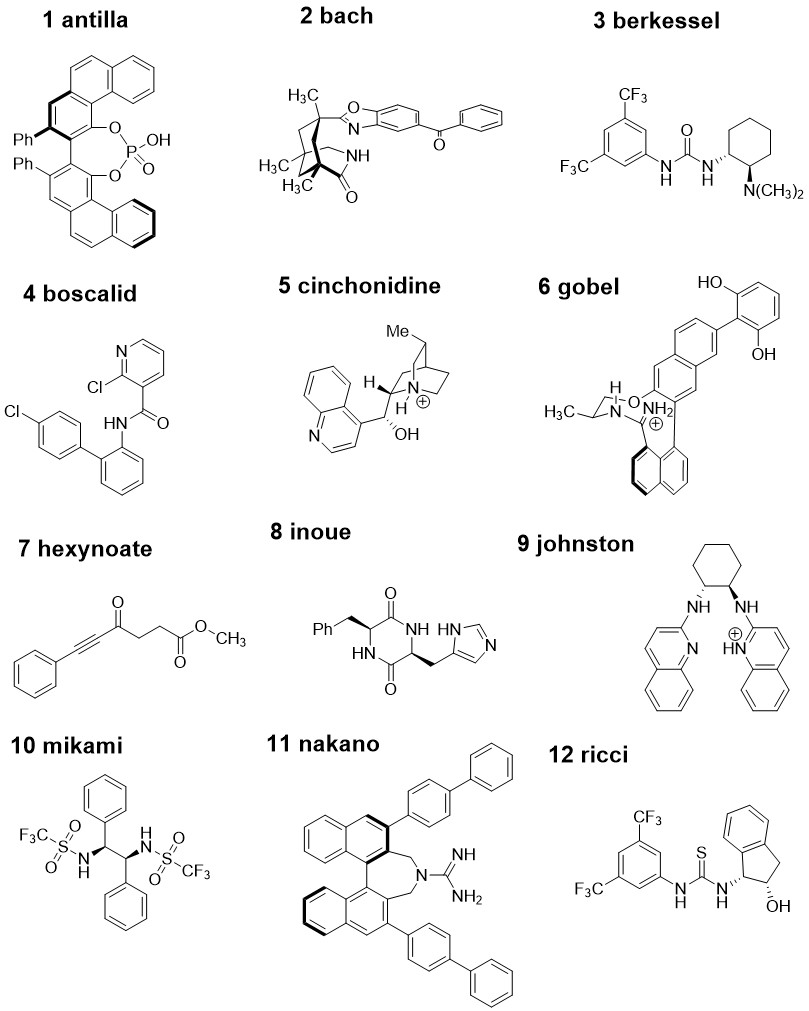
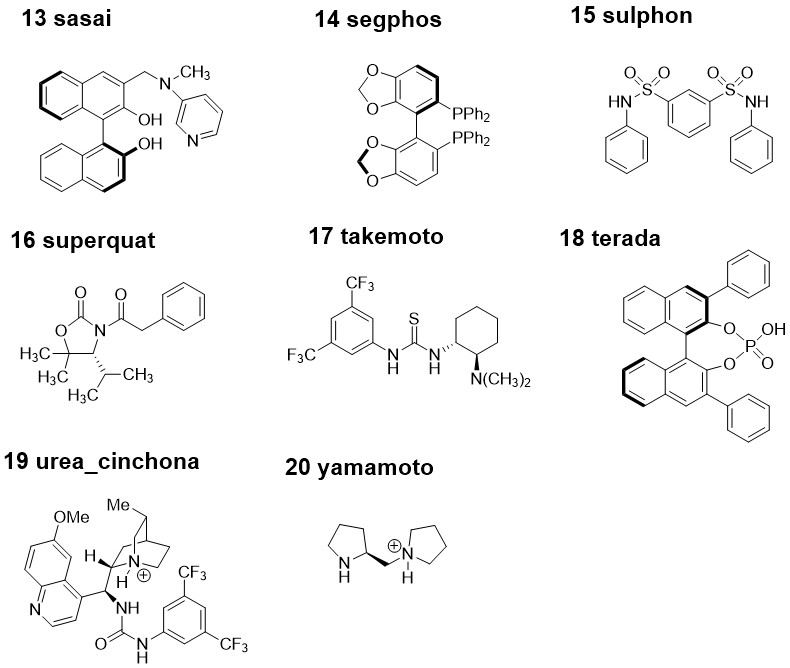
Each of the 20 molecules in the dataset was given a nickname that is used for the calculation files. The molecule numbers (used in the publication) and 2D molecular structures that these nicknames correspond to can be found from the images "molecule_names_and_structures_1.jpg" and "molecule_names_and_structures_2.jpg". Within each molecule directory, there is one directory for each force field that was able to conformationally search that molecule (format: <mol_name>/<mol_name_FF>). Within the <mol_name_FF> directories are the .mol2 files that correspond to the force field structures from the conformational searches. As well as the force field structures, the <mol_name_FF> directories each contain a "mo62x_6-31Gd" directory that contain the M06-2X/6-31G(d) optimised minima as Gaussian .out files and the M06-2X/def2-TZVPP//M06-2X/6-31G(d) single-point energy calculations as "_SPEopt.out" files. The "csvs" directory contains one .csv file for each molecule, each containing all of the force field and DFT energetic data for all conformers from each force field. The columns of each .csv data file are: conf: The conformer numbers from the searches FFE: The energies of the conformers accourding to the force fields Structure: The filenames of the DFT minima E_SPC: The single-point corrected DFT energy E: The uncorrected M06-2X/6-31G(d) energy ZPE: The zero-point energy H_SPC: The single-point corrected enthalpy T.S: The product of the temperature (298.15 K) and the entropy T.qh-S: The product of the temperature and the quasi-harmonic entropy G(T)_SPC: The single-point corrected Gibbs free energy qh-G(T)_SPC: The single-point corrected quasi-harmonic Gibbs free energy
Documentation Files
molecule_names … structures_1.jpg
image/jpeg (103kB)
Creative Commons: Attribution 4.0
molecule_names … structures_2.jpg
image/jpeg (72kB)
Creative Commons: Attribution 4.0
Funders
Engineering and Physical Sciences Research Council
https://doi.org/10.13039/501100000266
EPSRC Centre for Doctoral Training in Sustainable Chemical Technologies
EP/L016354/1
Engineering and Physical Sciences Research Council
https://doi.org/10.13039/501100000266
UKRI Centre for Doctoral Training in Accountable, Responsible and Transparent AI
EP/S023437/1
University of Bath
https://doi.org/10.13039/501100000835
Publication details
Publication date: 27 April 2022
by: University of Bath
Version: 1
DOI: https://doi.org/10.15125/BATH-01065
URL for this record: https://researchdata.bath.ac.uk/1065
Related papers and books
Lewis-Atwell, T., Townsend, P. A., and Grayson, M. N., 2022. Comparing the Performances of Force Fields in Conformational Searching of Hydrogen-Bond-Donating Catalysts. The Journal of Organic Chemistry, 87(9), 5703-5712. Available from: https://doi.org/10.1021/acs.joc.2c00066.
Contact information
Please contact the Research Data Service in the first instance for all matters concerning this item.
Contact person: Matthew Grayson
Faculty of Science
Chemistry

{kind=link}
{kind=link}